LE SYNDROME DE WOLFF PARKINSON WHITE:
Le syndrome de Wolff-Parkinson-White est une maladie où est présente, dès la naissance, une connexion électrique supplémentaire entre les oreillettes et les ventricules. La personne peut subir des épisodes de battements cardiaques très rapides.
- La plupart des patients ont des palpitations, et certains se sentent affaiblis ou essoufflés.
- Le diagnostic est basé sur une électrocardiographie.
- Les épisodes peuvent généralement être arrêtés par des manœuvres qui stimulent le nerf vague, ce qui ralentit le rythme cardiaque.
Le syndrome de Wolff-Parkinson-White est le plus fréquent des nombreux troubles engageant une voie de conduction supplémentaire (accessoire) entre les oreillettes et les ventricules. (Ces troubles sont appelés tachycardies supraventriculaires réciproques auriculo-ventriculaires.) Cette voie supplémentaire rend les arythmies rapides plus susceptibles de se produire. Bien que congénital, le syndrome de Wolff-Parkinson-White occasionne des arythmies qui se manifestent en général durant l’adolescence ou chez le jeune adulte. Cependant, les arythmies peuvent survenir au cours de la première année de vie ou après 60 ans.
Symptômes
Le syndrome de Wolff-Parkinson-White est une cause fréquente de tachycardie paroxystique supraventriculaire. Très rarement, ce syndrome provoque un rythme cardiaque extrêmement rapide et potentiellement mortel pendant la fibrillation auriculaire.
Si les nourrissons développent des arythmies dues à ce syndrome, ils peuvent devenir essoufflés ou léthargiques, perdre leur appétit, ou présenter des pulsations de thorax rapides et visibles. Une insuffisance cardiaque peut se développer.
En général, quand des adolescents ou de jeunes adultes dans la vingtaine subissent pour la première fois une arythmie due à ce syndrome, il s’agit d’un épisode de palpitations qui débute brutalement, souvent à l’occasion d’un effort physique. Il peut ne durer que quelques secondes ou persister pendant plusieurs heures. Dans la plupart des cas, le rythme cardiaque très rapide est très inconfortable et affligeante. Certaines personnes s’évanouissent.
Lorsque des épisodes de tachycardie paroxystique supraventriculaire due à un syndrome de Wolff-Parkinson-White surviennent chez des personnes plus âgées, ils provoquent en général plus de symptômes, comme un évanouissement, un essoufflement et une douleur thoracique.
Fibrillation auriculaire et syndrome de Wolff-Parkinson-White
La fibrillation auriculaire peut être particulièrement dangereuse en cas de syndrome de Wolff-Parkinson-White. La voie accessoire peut conduire les impulsions rapides vers les ventricules à un rythme beaucoup plus rapide que celui de la voie normale (à travers le nœud auriculo-ventriculaire). Il en résulte une fréquence ventriculaire extrêmement rapide et potentiellement mortelle. Le cœur est non seulement très peu efficace lorsqu’il bat aussi rapidement, mais ce rythme cardiaque extrêmement élevé peut également évoluer en une fibrillation ventriculaire, qui est mortelle si elle n’est pas rapidement traitée.
Diagnostic :
- Électrocardiographie
Comme le syndrome de Wolff-Parkinson-White modifie le schéma d’activation électrique dans le cœur, il peut être diagnostiqué par une électrocardiographie (ECG), qui enregistre l’activité électrique du cœur.
Traitement :
- Manœuvres et médicaments pour convertir le rythme cardiaque
- Parfois ablation par radiofréquence
Les épisodes de tachycardie paroxystique supraventriculaire dus au syndrome de Wolff-Parkinson-White peuvent souvent être arrêtés par l’une des manœuvres qui stimulent le nerf vague et donc ralentissent le rythme cardiaque. Ces manœuvres sont plus efficaces si elles sont exécutées peu de temps après le début de l’arythmie. Lorsqu’elles sont inefficaces, des médicaments, comme le vérapamil et l’adénosine, sont en général administrés par voie intraveineuse pour arrêter l’arythmie. Des anti-arythmiques peuvent alors être pris indéfiniment pour prévenir de nouveaux épisodes de rythme cardiaque rapide.
Chez les nourrissons et les enfants de moins de 10 ans, la digoxine peut être administrée pour supprimer les épisodes de tachycardie paroxystique supraventriculaire dus au syndrome de Wolff-Parkinson-White. Cependant, les adultes atteints de ce syndrome ne doivent pas prendre de digoxine car elle peut faciliter la conduction par la voie accessoire et augmenter le risque qu’une fibrillation auriculaire induise une fibrillation ventriculaire. L’administration de la digoxine est, pour cette raison, en général arrêtée avant la puberté chez les personnes qui présentent ce syndrome.
Ablation par radiofréquence
La destruction de la voie de conduction accessoire par ablation par radiofréquence (distribution d’énergie à une fréquence spécifique à l’aide d’un cathéter à électrode introduit dans le cœur) est réussie chez plus de 95 % des patients. Le risque de décès pendant la procédure est inférieur à 1 sur 1 000. L’ablation par radiofréquence est particulièrement utile chez les jeunes qui devraient autrement prendre des anti-arythmiques à vie.
LES CANALOPATHIES CARDIAQUES:
Les canalopathies cardiaques sont des anomalies génétiques des protéines des cellules cardiaques qui contrôlent l’activité électrique du cœur et peuvent donc provoquer des troubles du rythme.
La plupart des personnes atteintes de canalopathies cardiaques n’ont pas d’autres maladies cardiaques, telles qu’une crise cardiaque ou une maladie des valvules cardiaques, mais elles sont porteuses de mutations des gènes qui déterminent la composition ou la régulation des pores des membranes cardiaques (canaux) et sont prédisposées à des anomalies du rythme cardiaque. Les canalopathies les plus fréquentes provoquent:
- Syndrome du QT long
D’autres canalopathies cardiaques plus rares comprennent :
- Syndrome du QT court
- Syndrome de l’onde J
- Tachycardie ventriculaire polymorphe catécholaminergique (TVPC)
- Syndrome de Brugada
Les anomalies électriques causent parfois une tachycardie ventriculaire ou une fibrillation ventriculaire.
Symptômes :
Certaines personnes n’ont aucun symptôme, mais nombre d’entre elles s’évanouissent à cause d’une tachycardie ventriculaire. Celles qui souffrent de fibrillation ventriculaire subissent un arrêt cardiaque soudain. Ils peuvent être déclenchés par une fièvre ou un traitement avec certains médicaments, dont certains anti-arythmiques et antidépresseurs.
Diagnostic:
- Électrocardiographie
Le diagnostic est basé sur une électrocardiographie (ECG). Mais il arrive que le schéma des anomalies ECG soit moins clair. Dans ces cas, les médecins peuvent essayer de provoquer des troubles du rythme cardiaque avec un médicament ou une activité physique, ce qui leur permet de réaliser un diagnostic.
Traitement:
En général, on utilise un Défibrillateur Automatique Implantable (DAI), un petit dispositif capable de détecter une arythmie et d’administrer un choc pour la corriger. Cette procédure est analogue à l’implantation d’un stimulateur cardiaque artificiel.
LE SYNDROME DU QT LONG:
Les torsades de pointes sont une forme particulière de tachycardie ventriculaire polymorphe survenant chez le patient présentant un allongement pathologique de l’intervalle QT. La tachycardie est caractérisée par une succession de QRS d’amplitude variable et dont la polarité semble osciller autour de la ligne isoélectrique de l’ECG. Cette forme de troubles du rythme peut s’arrêter spontanément ou dégénérer en fibrillation ventriculaire. Les torsades de pointes entraînent des risques d’instabilité hémodynamique sévères et souvent de décès. Le diagnostic repose sur l’ECG. Le traitement est repose sur l’administration de Mg IV, sur différentes mesures tendant à raccourcir le QT et sur le choc électrique externe en cas de fibrillation ventriculaire.
L’intervalle QT long responsable de torsades de pointes peut être congénital ou médicamenteux. Un allongement de l’intervalle QT prédispose aux troubles du rythme par l’allongement excessif de la repolarisation ventriculaire qui induit des post-dépolarisations précoces et une dispersion spatiale des périodes réfractaires des ventricules.
Syndrome du QT long congénital :
Au moins 10 formes différentes de syndrome du QT long congénital ont été décrites. La plupart des cas appartiennent aux 3 premiers sous-groupes:
Syndrome du QT long type 1 (LQT1), provoqué par une mutation avec perte de fonction du gène KCNQ1, qui encode le courant K I sensibles aux catécholamines IKs
Syndrome du QT long type 2 (LQT2), provoqué par une mutation avec perte de fonction du gène HERG, qui encode un autre canal K cardiaque (IKr)
Syndrome du QT long type 3 (LQT3) est causé par une mutation du gène SCN5A, qui perturbe l’inactivation rapide du canal cardiaque Na (INa)
Ces formes sont héréditaires à transmission autosomique dominante à pénétrance incomplète et, par le passé, étaient appelées syndrome de Romano-Ward. Chez de rares patients qui présentent 2 copies anormales de l’anomalie génétique (en particulier LQT1), le trouble est associé à une surdité congénitale et par le passé, était appelé syndrome de Jervell et Lange-Nielsen. Les patients qui présentent un syndrome du QT long sont sujets aux syncopes récidivantes secondaires à des torsades de pointe et sont à risque de mort subite secondaire à une torsade de pointe dégénérant en fibrillation ventriculaire.
Syndrome du QT long induit par les médicaments :
La cause la plus fréquente de torsades de pointes VT de la tachycardie ventriculaire est généralement la prise d’antiarythmiques de la classe Ia, Ic ou III. D’autres médicaments qui peuvent induire des torsades de pointes sont les antidépresseurs tricycliques, les phénothiazines et certains antiviraux et antifongiques.
Symptomatologie :
Le patient se présente souvent avec une syncope, du fait du rythme ventriculaire sous-jacent très rapide entre (200 et 250 battements/minute). Les palpitations sont également un symptôme fréquent chez le patient conscient. Parfois, un QT long est détecté après les manœuvres de réanimation.
Diagnostic :
ECG
Le diagnostic repose sur l’ECG qui révèle un axe QRS ondulant avec une polarité des complexes oscillant continuellement autour de la ligne de base ( Torsades de pointes.). L’ECG intercritique permet d’observer l’allongement pathologique du QT après correction en fonction de la fréquence cardiaque (QTc). Les valeurs normales sont en moyenne d’environ 0,44 s, bien qu’elles varient selon les individus et le sexe. Des antécédents familiaux peuvent faire évoquer un syndrome congénital.
Torsades de pointes.

Calculateur clinique : Intervalle QT corrigé
Traitement :
Habituellement, cardioversion (choc électrique) externe non synchronisée
Parfois, sulfate de Mg (MgSO4) IV
Un épisode de torsades de pointes aigu soutenu s’accompagnant d’une instabilité hémodynamique est traité par cardioversion électrique externe débutée à 100 joules. Cependant, une récidive précoce est de règle. Les patients répondent souvent au Mg, habituellement MgSO4 2 g IV en 1 à 2 min. Si ce traitement est inefficace, un 2e bolus est administré en 5 à 10 min suivi d’une perfusion continue de 3 à 20 mg/min chez un patient qui ne présente pas d’insuffisance rénale. La lidocaïne (un anti-arythmique de classe Ib) raccourcit l’intervalle QT et peut être efficace en particulier lorsque les torsades de pointes sont secondaires à la prise de médicaments. Les molécules des classes Ia, Ic et III sont contre-indiquées.
Si un médicament est la cause des torsades de pointes, il est arrêté, mais jusqu’à son élimination complète, le patient qui présente de fréquents ou longs épisodes de torsades de pointes sera traité pour raccourcir l’intervalle QT. L’augmentation de la fréquence cardiaque raccourcissant le QT, une stimulation cardiaque temporaire et/ou l’administration d’isoprotérénol IV est souvent efficace.
Un traitement à long terme est requis en cas de syndrome du QT long congénital. Les thérapeutiques de choix comprennent les β-bloqueurs, l’implantation d’un pacemaker ou d’un défibrillateur implantable ou une association de ces traitements. Un enregistrement ECG doit être réalisé chez tous les membres de la famille.
Les patients qui présentent un syndrome du QT long congénital doivent clairement éviter les médicaments qui prolongent l’intervalle QT et les patients qui présentent des symptômes liés à l’exercice (habituellement LQT1 ou LQT2) doivent éviter les exercices physiques. Les options thérapeutiques comprennent les β-bloqueurs, la stimulation pour maintenir une fréquence cardiaque plus rapide (qui raccourcit l’intervalle QT), et le cardioverteur-défibrillateur implantable, seuls ou en association. Les lignes directrices actuelles recommandent un cardioverteur-défibrillateur implantable après réanimation d’un arrêt cardiaque et des syncopes malgré un traitement β-bloqueur.
Points clés:
L’intervalle QT long responsable de torsades de pointes peut être congénital ou médicamenteux.
Le traitement immédiat des torsades est la cardioversion non synchronisés en commençant par 100 joules, bien que certains patients réagissent positivement au MgSO4 2 g IV en 1 à 2 min.
Les thérapeutiques de choix comprennent les β-bloqueurs, l’implantation d’un pacemaker permanent ou d’un cardioverteur-défibrillateur implantable ou une association de ces traitements.
Un enregistrement ECG doit être réalisé chez tous les membres de la famille.
LES EXTRASYSTOLES VENTRICULAIRES:
Une extrasystole ventriculaire est un battement cardiaque supplémentaire dû à une activation électrique anormale, dont l’origine se situe dans les ventricules et qui survient avant un battement cardiaque normal.
Le symptôme principal est la perception d’un battement manqué.
Le diagnostic est basé sur une électrocardiographie.
Le traitement qui consiste à éviter ce qui déclenche ces battements, comme le stress, la caféine, et l’alcool, est généralement suffisant.
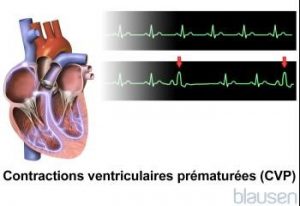
Les extrasystoles ventriculaires sont fréquentes, notamment chez les personnes âgées Ces arythmies peuvent être provoquées par un stress physique ou émotionnel, la consommation de caféine (dans les boissons ou la nourriture) ou d’alcool, ou par des remèdes pour le rhume ou le rhume des foins contenant des médicaments qui stimulent le cœur, comme la pseudo-éphédrine. Des causes additionnelles comprennent les maladies des artères coronaires (surtout pendant ou peu après une crise cardiaque) et les troubles qui induisent la dilatation ventriculaire, tels que l’insuffisance cardiaque et les maladies des valvules cardiaques.
Symptômes:
Les extrasystoles ventriculaires isolées diminuent peu l’efficacité contractile cardiaque si elles ne sont pas trop fréquentes, et sont donc habituellement asymptomatiques. Le symptôme principal est la perception d’un battement fort ou manquant. Les extrasystoles ventriculaires ne sont pas dangereuses chez les personnes qui ne présentent pas de maladie cardiaque. Cependant, quand elles sont fréquentes chez des personnes souffrant de maladie organique du cœur (comme une maladie des valvules cardiaques ou un infarctus du myocarde), elles peuvent être annonciatrices d’arythmies plus dangereuses, comme une tachycardie ventriculaire ou une fibrillation ventriculaire, susceptibles de causer une mort subite.
Diagnostic:
Électrocardiographie
L’électrocardiographie (ECG) est utilisée pour diagnostiquer les extrasystoles ventriculaires.
Traitement:
-Modifications du mode de vie
-Parfois bêta-bloquants
Les personnes qui sont par ailleurs en bonne santé n’ont besoin d’aucun traitement, si ce n’est de diminuer le stress et d’arrêter de consommer de la caféine, de l’alcool ou des médicaments en vente libre contre le rhume ou le rhume des foins qui contiennent des substances qui stimulent le cœur. Un traitement pharmacologique est rarement utilisé car le risque d’effets secondaires dus aux médicaments surpasse généralement ses bénéfices. L’exception concerne les personnes qui ont subi un infarctus du myocarde récent ou qui souffrent d’une insuffisance cardiaque qui cause des symptômes. La survie de ces personnes est améliorée avec un traitement par bêta-bloquants.