LES CARDIOMYOPATHIES
La cardiomyopathie est une altération progressive de la structure et de la fonction des parois musculaires des cavités cardiaques.
Types de cardiomyopathie :
Il y a trois principaux types de cardiomyopathies : dilatée, hypertrophique et restrictive.
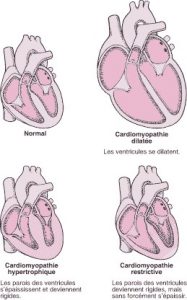
La cardiomyopathie peut être causée par de nombreux troubles, elle peut également n’avoir aucune cause identifiable. Les trois principaux types de cardiomyopathies, qui peuvent se chevaucher, sont les cardiomyopathies dilatée, hypertrophique et restrictive. Les cardiomyopathies causent souvent des symptômes d’insuffisance cardiaque. Certaines cardiomyopathies peuvent également causer une douleur thoracique, des évanouissements, ou une mort subite.
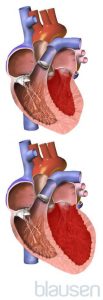
Cœur normal vs hypertrophié
LA CARDIOMYOPATHIE HYPERTROPHIQUE:
La cardiomyopathie hypertrophique regroupe des troubles cardiaques qui provoquent l’épaississement (hypertrophie) et le raidissement des parois des ventricules.
Les cas de cardiomyopathie hypertrophique sont généralement causés par une anomalie génétique héritée.
Les patients subissent des syncopes, des douleurs thoraciques, des essoufflements, et perçoivent des battements cardiaques irréguliers.
Les médecins basent leur diagnostic sur les résultats d’un examen clinique, mais ils utilisent l’échocardiographie ou l’imagerie par résonance magnétique pour le confirmer.
Des médicaments qui réduisent la force des contractions du cœur sont administrés.
La cardiomyopathie hypertrophique est une cause fréquente de mort subite chez les jeunes athlètes. Environ 1 personne sur 500 est affectée.
Causes :
La cardiomyopathie hypertrophique est presque toujours causée par une anomalie génétique héritée. Très rarement, les personnes développent une cardiomyopathie hypertrophique quand elles présentent des troubles comme une acromégalie (croissance excessive due à la surproduction d’hormone de croissance, généralement par une hypophyse bénigne), un phéochromocytome (une tumeur qui produit un excès de l’hormone épinéphrine), ou une neurofibromatose (un trouble génétique dans lequel de nombreuses croissances molles et charnues de tissu nerveux apparaissent sous la peau et dans d’autres parties du corps).
Les parois épaisses et rigides des ventricules ne se relaxent pas correctement pour permettre aux cavités cardiaques de se remplir de sang. Cette difficulté devient plus sévère quand le cœur bat rapidement (durant l’exercice physique par exemple) car le cœur a encore moins de temps pour se remplir. Comme le cœur ne se remplit pas correctement, il pompe moins de sang avec chaque battement. La paroi épaissie du cœur interfère également parfois avec le flux sanguin hors du cœur. Cette variation est appelée cardiomyopathie obstructive hypertrophique.
En raison de l’épaississement des parois des ventricules, la valvule mitrale (qui s’ouvre entre l’oreillette gauche et le ventricule gauche) peut être incapable de se fermer normalement, entraînant le reflux d’une petite quantité de sang dans l’oreillette gauche. Cette fuite de valvule et les parois ventriculaires élargies causent typiquement des sons cardiaques anormaux (souffles cardiaques).
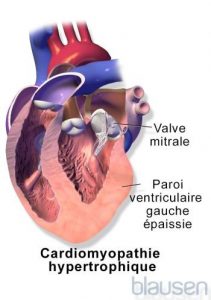
Cardiomyopathie hypertrophique
Symptômes :
Les symptômes sont extrêmement variables, mais quand ils se produisent, ils se développent généralement quand les individus sont âgés de 20 à 40 ans. Les symptômes apparaissent durant l’effort et comprennent des syncopes (syncope), des douleurs thoraciques, des essoufflements, et une sensation de battements cardiaques irréguliers (palpitations). Les syncopes sont généralement subites sans symptômes avertisseurs. Les syncopes ou même une mort subite peuvent être les premiers signes qu’une personne est atteinte de ce trouble.
Diagnostic :
Les médecins déterminent généralement le diagnostic de cardiomyopathie hypertrophique en fonction des symptômes du patient, des résultats d’un examen clinique, d’une électrocardiographie (ECG), et d’une radiographie des poumons. Les sons cardiaques et les souffles entendus à l’aide d’un stéthoscope peuvent être utiles. L’échocardiographie est la meilleure façon de confirmer le diagnostic, mais l’imagerie par résonance magnétique (IRM) est également utile car elle peut fournir des informations plus détaillées. Le cathétérisme cardiaque, une procédure invasive, est pratiqué pour mesurer les pressions dans les cavités cardiaques seulement si une intervention chirurgicale est envisagée.
Comme la cardiomyopathie hypertrophique est généralement causée par une mutation génique, des tests génétiques peuvent également être réalisés.
Pronostic :
Chaque année, environ 1 à 3 % des adultes atteints de cardiomyopathie hypertrophique y succombent. Les enfants atteints de cardiomyopathie hypertrophique risquent plus de mourir. Les autres facteurs de risque sont la gravité de l’épaississement du muscle cardiaque, la présence de rythmes ventriculaires rapides, un effort physique excessif soudain, et la non-observance des recommandations des médecins. La mort est généralement soudaine et vraisemblablement due à un trouble du rythme cardiaque. La mort due à une insuffisance cardiaque chronique est moins fréquente. Les personnes qui apprennent qu’elles ont hérité de ce trouble peuvent demander une consultation génétique si elles envisagent de fonder une famille car le risque qu’elles transmettent ce trouble à leurs enfants est de 50 %. Les membres de la famille des personnes présentant ce trouble hérité peuvent également envisager des tests génétiques.
Traitement :
Si possible, les médecins traitent la cause sous-jacente.
Le traitement de la cardiomyopathie hypertrophique vise principalement à réduire la résistance du cœur à se remplir de sang entre les battements. Les bêta-bloquants et l’inhibiteur calcique vérapamil, administrés seuls ou en association, constituent le traitement principal. Les deux médicaments réduisent la contractilité du muscle cardiaque pour que le cœur se contracte moins vigoureusement. Par conséquent, le cœur se remplit mieux et, si le muscle hypertrophié bloquait le flux sanguin, le sang peut maintenant être plus facilement éjecté par le cœur. Les bêta-bloquants et le vérapamil ralentissent aussi la fréquence cardiaque, ce qui donne plus de temps au cœur pour se remplir. La disopyramide, un médicament qui diminue la force des contractions cardiaques, est aussi utilisée.
Une intervention chirurgicale visant à enlever une partie du muscle cardiaque épaissi (myectomie) peut améliorer le flux sanguin provenant du cœur, mais une telle intervention est seulement réalisée quand les symptômes sont débilitants malgré le traitement pharmacologique. La myectomie peut soulager les symptômes mais ne diminue pas le risque de mort. L’ablation à l’alcool (la destruction contrôlée d’une petite partie du muscle cardiaque) est de plus en plus pratiquée chez certains patients pour améliorer le flux sanguin provenant du cœur car elle peut se faire par cathétérisme cardiaque. Bien que le cathétérisme cardiaque soit une procédure invasive dans laquelle un cathéter est introduit dans le cœur, elle comporte moins de risques qu’une intervention chirurgicale. Toutefois, quand la myectomie est réalisée dans un hôpital très expérimenté dans cette procédure, les résultats à long terme sont excellents.
Dans un sous-groupe de personnes souffrant de cardiomyopathie hypertrophique, un risque élevé de mort subite existe chez celles qui présentent un épaississement plus sévère du muscle, notamment dans la paroi qui sépare les cavités cardiaques l’une de l’autre (septum cardiaque). Les médecins peuvent recommander un défibrillateur automatique implantable pour ces personnes.
LA CARDIOMYOPATHIE DILATÉE:
La cardiomyopathie dilatée (congestive) est un groupe de troubles du muscle cardiaque dans lesquels les ventricules se dilatent sans pouvoir pomper une quantité suffisante de sang dans le corps, ce qui provoque une insuffisance cardiaque.
La coronaropathie, des infections virales, et certains troubles hormonaux sont des causes fréquentes de cardiomyopathie dilatée.
L’essoufflement et la fatigue en sont souvent les premiers symptômes.
L’électrocardiographie, l’échocardiographie, l’imagerie par résonance magnétique, et des analyses sanguines sont utilisées pour diagnostiquer une cardiomyopathie dilatée.
Les médecins tentent généralement de traiter la cause de cette cardiomyopathie en prescrivant des médicaments.
La cardiomyopathie dilatée peut se manifester à n’importe quel âge, mais elle est plus fréquente entre 20 et 60 ans. Environ 10 % des personnes présentant une cardiomyopathie dilatée ont plus de 65 ans. Ce trouble est environ 3 fois plus fréquent chez les hommes que chez les femmes et 3 fois plus fréquent chez les Noirs que chez les Blancs. Environ 5 à 8 personnes sur 100 000 développent ce trouble chaque année.
Causes :
En Amérique du Nord, la cause identifiable la plus fréquente de cardiomyopathie dilatée est une coronaropathie extensive. Une telle coronaropathie a pour conséquence une irrigation inadéquate du muscle cardiaque, ce qui entraîne la lésion permanente et la nécrose du muscle cardiaque. Par conséquent, le cœur ne peut pas pomper aussi vigoureusement. Le muscle cardiaque nécrosé est remplacé par du tissu fibreux (cicatriciel). La partie du cœur qui reste intacte se distend alors et s’épaissit (hypertrophies) pour compenser la capacité de pompage perdue. Plus le muscle cardiaque est étiré, le plus vigoureusement il se contracte ou pompe, mais seulement jusqu’à un certain point. Au-delà de ce point, l’épaississement et l’extension ne compensent pas adéquatement, entraînant une cardiomyopathie dilatée avec insuffisance cardiaque.
La cardiomyopathie dilatée peut aussi être due à une inflammation aiguë du muscle cardiaque (myocardite) résultant d’une infection virale. Ce trouble est appelé cardiomyopathie virale. En Amérique du Nord, l’infection par le virus coxsackie B est la cause la plus fréquente de cardiomyopathie virale. Dans d’autres parties du monde, elle est causée plus fréquemment par d’autres infections virales. Le virus infecte et fragilise le muscle cardiaque. Comme dans la coronaropathie, le cœur fragilisé se distend pour compenser, provoquant une cardiomyopathie dilatée et souvent une insuffisance cardiaque. La cardiomyopathie dilatée parfois est secondaire à une infection bactérienne.
Les autres causes de cardiomyopathie dilatée sont certains troubles hormonaux chroniques, comme un diabète de longue date mal contrôlé ou une maladie thyroïdienne, une hypertension artérielle, une obésité morbide, ou un rythme cardiaque rapide persistant. La cardiomyopathie dilatée peut aussi être causée par la prise de certaines substances dont l’alcool (quand la consommation est excessive et associée à une malnutrition), la cocaïne, les antidépresseurs, les antipsychotiques les plus récents, ainsi que certains médicaments utilisés en chimiothérapie. Les causes rares de cardiomyopathie dilatée sont la grossesse, une surcharge en fer et des maladies du tissu conjonctif comme la polyarthrite rhumatoïde. Les facteurs génétiques jouent un rôle dans 20 à 35 % des cas. Quand aucune cause spécifique ne peut être identifiée, on parle de cardiomyopathie dilatée idiopathique.
Symptômes :
En général, les premiers symptômes de cardiomyopathie dilatée sont un essoufflement à l’effort et une tendance à la fatigue. Ils résultent d’une baisse de la capacité de pompage du cœur, appelée insuffisance cardiaque. Quand la cardiomyopathie résulte d’une infection, les premiers symptômes peuvent être une fièvre subite et des symptômes de grippe. Quelle qu’en soit la cause, si le cœur est suffisamment endommagé, la fréquence cardiaque finit par s’accélérer, la pression artérielle est normale ou basse, on observe une rétention de liquide dans les jambes et l’abdomen, et les poumons se remplissent de liquide.
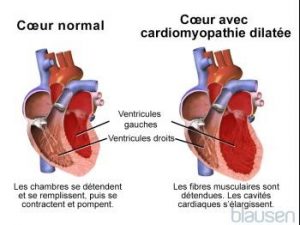
Cardiomyopathie dilatée
Comme le cœur est dilaté, il arrive que les valvules cardiaques soient incapables de se fermer normalement et permettent souvent au sang de refluer dans la cavité cardiaque au lieu de s’écouler dans la chambre ou dans le vaisseau sanguin suivant(e). Les valvules les plus souvent affectées sont la valvule mitrale, située entre l’oreillette gauche (cavité cardiaque supérieure) et le ventricule gauche (cavité cardiaque inférieure), et la valvule tricuspide, située entre l’oreillette droite et le ventricule droit. La fuite provoque un souffle qui peut être entendu par les médecins avec un stéthoscope. Le dommage occasionné au muscle cardiaque et sa dilatation peuvent provoquer des anomalies du rythme cardiaque (arythmies), qui peuvent causer une sensation de battements de cœur irréguliers (palpitations) ou entraîner la mort. L’insuffisance valvulaire et les troubles du rythme cardiaque peuvent altérer encore plus la capacité de pompage du cœur.
Le sang peut stagner dans le cœur dilaté, augmentant le risque de formation de caillots sur les parois des chambres cardiaques. Ces caillots peuvent se fragmenter (en emboles), passer du cœur aux vaisseaux sanguins ailleurs dans le corps, et les obstruer, endommageant l’organe qu’ils irriguent. Si l’apport de sang au cerveau est bloqué, un accident vasculaire cérébral peut se produire.
Diagnostic :
Le diagnostic repose sur les symptômes, sur les résultats d’un examen clinique, et sur d’autres tests. Une électrocardiographie (ECG) peut détecter les anomalies de l’activité électrique du cœur. Toutefois, ces anomalies ne constituent pas une preuve suffisante pour un diagnostic. L’échocardiographie, qui utilise des ultrasons pour produire une image du cœur, est la procédure la plus utile car elle peut indiquer la taille et la capacité de pompage du cœur. L’imagerie par résonance magnétique cardiaque, qui produit des images très détaillées du cœur, est utilisée le plus souvent pour confirmer le diagnostic (et parfois pour identifier la cause).
Si le diagnostic reste douteux, un cathétérisme cardiaque, une procédure invasive dans laquelle un cathéter est introduit dans le cœur, peut fournir un complément d’information sur la capacité de pompage du cœur. Le cathétérisme cardiaque permet également aux médecins de mesurer les pressions dans les cavités cardiaques et de déterminer l’étendue de la coronaropathie. Pendant le cathétérisme, un échantillon de tissu peut aussi être prélevé pour être examiné au microscope (biopsie). La biopsie peut parfois révéler des changements microscopiques caractéristiques de certaines maladies qui causent une cardiomyopathie dilatée (comme une infection virale récente) et confirmer le diagnostic. Cependant, les résultats d’une biopsie ne sont généralement pas assez spécifiques pour permettre un diagnostic.
Pronostic :
Le pronostic varie considérablement en fonction de nombreux facteurs. En général, le pronostic s’aggrave à mesure que le cœur se dilate et fonctionne moins bien. Des anomalies du rythme cardiaque suggèrent également un pronostic plus grave. En général, la survie des hommes est inférieure de moitié à celle des femmes, et la survie des Noirs est inférieure de moitié à celle des Blancs. Environ 40 à 50 % des décès sont subits, résultant probablement d’une anomalie du rythme cardiaque ou d’un embole qui bloque le flux sanguin dans une zone critique. Parmi les autres facteurs d’explication possibles figurent aussi la cause et la sévérité de la cardiomyopathie, l’âge de la personne et sa capacité à observer les conseils de son médecin, l’accès à un traitement spécialisé, le sel dans l’alimentation et les consultations ambulatoires. Le pronostic général s’est toutefois amélioré au cours des dernières années avec les traitements actuels, notamment avec l’introduction de défibrillateurs cardioverteurs implantables, de la thérapie de resynchronisation cardiaque, et d’autres interventions.
Traitement :
Si possible, les médecins traitent la cause sous-jacente.
Les traitements habituels consistent à éviter le stress, limiter le sel dans l’alimentation, et à observer des périodes de repos qui réduisent l’effort appliqué sur le cœur, en particulier quand la cardiomyopathie est aiguë ou sévère.
Des médicaments comme les inhibiteurs de l’enzyme de conversion de l’angiotensine (ECA), les antagonistes des récepteurs de l’angiotensine II, les agents bêta-bloquants, les antagonistes de l’aldostérone (spironolactone ou éplérénone), et la digoxine à faible dose, améliorent la capacité de pompage du cœur, prolongent la vie, et réduisent les symptômes persistants. Des diurétiques sont utilisés pour réduire l’excès de liquide dans les poumons et les symptômes d’œdème attribuables à une rétention de liquides, mais ils ne prolongent pas la vie.
Des anti-arythmiques peuvent être prescrits pour traiter les anomalies du rythme cardiaque. La plupart de ces médicaments sont initialement prescrits en petites doses. Les doses sont augmentées petit à petit car, si la dose est excessive, un anti-arythmique peut aggraver les anomalies du rythme cardiaque ou déprimer la capacité de pompage. Certaines personnes présentent une anomalie de la conduction électrique cardiaque, ce qui peut être traité par un stimulateur cardiaque qui stimule d’abord les oreillettes et puis les ventricules (thérapie de resynchronisation cardiaque). Ce type de stimulation, utilisé pour la bonne personne, permet de rétablir le rythme normal de contraction du cœur et d’améliorer sa fonction. Les médecins peuvent également envisager un défibrillateur cardioverteur implantable pour les patients atteints d’un dysfonctionnement cardiaque persistant et à risque accru de mort subite.
Quelle que soit la cause de la cardiomyopathie dilatée, les médecins peuvent prescrire des anticoagulants, comme la warfarine ou l’aspirine, pour éviter les caillots sanguins qui peuvent se former sur les parois des cavités cardiaques de ventricules très dilatés qui se contractent mal.
Si la cause spécifique de la cardiomyopathie dilatée ne peut pas être traitée, l’insuffisance cardiaque résultant d’une cardiomyopathie dilatée est progressive et éventuellement mortelle. À cause de ce mauvais pronostic, la cardiomyopathie dilatée est la raison la plus fréquente pour une greffe de cœur ou un dispositif d’assistance circulatoire mécanique. Une greffe cardiaque réussie guérit le trouble mais présente des complications et des limitations.
LA CARDIOMYOPATHIE RESTRICTIVE:
La cardiomyopathie restrictive (infiltrante) regroupe des troubles cardiaques qui provoquent un raidissement des parois des ventricules, mais pas nécessairement leur épaississement, et résistent au remplissage normal du cœur par le sang entre les battements.
La cardiomyopathie restrictive peut se produire quand le muscle cardiaque est graduellement infiltré ou remplacé par du tissu cicatriciel ou quand des substances anormales s’accumulent dans le muscle cardiaque.
Un essoufflement, une accumulation de liquide dans les tissus, des anomalies du rythme cardiaque, et une perception des battements cardiaques sont des symptômes fréquents.
Le diagnostic repose sur les résultats d’un examen clinique, d’une électrocardiographie (ECG), d’une échocardiographie, et d’un cathétérisme cardiaque.
Le traitement n’est pas toujours utile, bien que les médecins puissent parfois traiter la cause.
La forme de cardiomyopathie la moins fréquente, la cardiomyopathie restrictive, a de nombreuses caractéristiques en commun avec la cardiomyopathie hypertrophique. Sa cause est généralement inconnue.
Il y a deux principaux types de cardiomyopathie restrictive.
Le muscle cardiaque est remplacé progressivement par du tissu cicatriciel.
Des substances anormales s’accumulent ou s’infiltrent dans le muscle cardiaque.
Une cicatrisation peut résulter d’une lésion due à une radiothérapie pratiquée pour traiter une tumeur thoracique coexistante.
Si le corps contient trop de fer, par exemple, ce dernier peut s’accumuler dans le muscle cardiaque, ce qui est le cas chez les personnes qui présentent une surcharge en fer (hémochromatose). Des éosinophiles, un type de cellule sanguine, peuvent infiltrer le muscle cardiaque chez les personnes atteintes du syndrome hyperéosinophilique qui est plus prévalant dans les régions tropicales. L’amyloïde, une protéine inhabituelle normalement présente dans l’organisme, peut s’accumuler dans le muscle cardiaque et dans d’autres tissus et causer une amyloïdose. L’amyloïdose est plus fréquente chez les personnes âgées. D’autres exemples sont les tumeurs et le tissu granulomateux (accumulation anormale de globules blancs qui se forment en réponse à une inflammation chronique) qui se développent, par exemple, chez les personnes présentant une sarcoïdose.
Une forme congénitale de cardiomyopathie restrictive est observée chez les nourrissons souffrant de fibroélastose endocardique. Ce trouble rare est caractérisé par la présence d’une couche épaissie de tissu fibreux dans le ventricule gauche.
Symptômes :
La cardiomyopathie restrictive provoque une insuffisance cardiaque avec un essoufflement durant l’effort et en position allongée, ainsi qu’une accumulation de liquide dans les tissus (œdème). Les douleurs thoraciques et les évanouissements (syncope) sont moins fréquents que dans la cardiomyopathie hypertrophique, par contre, les anomalies du rythme cardiaque (arythmies) sont fréquentes. La fatigue est également possible. En général, les symptômes ne se manifestent pas au repos car, en cas de cardiomyopathie restrictive, le cœur peut fournir au corps un apport suffisant en sang et en oxygène au repos, bien que le cœur rigide résiste au remplissage par le sang. Des symptômes se manifestent à l’effort, lorsque le cœur rigide ne peut pas pomper une quantité suffisante de sang pour répondre aux besoins accrus du corps en sang et en oxygène.
Diagnostic :
La cardiomyopathie restrictive est l’une des causes potentielles examinées en cas d’insuffisance cardiaque. Le diagnostic repose largement sur les résultats combinés d’un examen clinique, d’une électrocardiographie (ECG), d’une radiographie des poumons, et d’une échocardiographie. L’ECG peut généralement détecter des anomalies de l’activité électrique du cœur mais celles-ci ne sont pas suffisamment spécifiques pour permettre un diagnostic. L’échocardiographie montre que les oreillettes sont dilatées et que le cœur ne fonctionne correctement que quand il se contracte (durant la systole). L’imagerie par résonance magnétique (IRM) peut détecter une texture anormale du muscle cardiaque due à l’accumulation ou à l’infiltration de substances anormales comme le fer et l’amyloïde. Bien que cette procédure ne soit pas souvent nécessaire, les médecins pratiquent parfois un cathétérisme cardiaque pour mesurer les pressions dans les cavités cardiaques et prélever un échantillon du muscle cardiaque pour l’examiner au microscope (biopsie), ce qui peut leur permettre d’identifier l’infiltration d’une substance. Plus d’une fois sur deux, la cause précise de la cardiomyopathie restrictive n’est pas déterminée (cardiomyopathie restrictive idiopathique).
Pronostic et traitement :
La survie varie selon la cause. Mais, en moyenne, la survie des personnes souffrant de cardiomyopathie restrictive idiopathique est d’environ 7 ans.
Pour la plupart des patients, un traitement n’est pas très utile. Les diurétiques, par exemple, généralement prescrits pour traiter une insuffisance cardiaque, peuvent aider les personnes souffrant d’un gonflement désagréable des jambes ou d’un essoufflement mais réduisent aussi la quantité de sang qui pénètre dans le cœur, ce qui peut aggraver la cardiomyopathie restrictive au lieu de l’améliorer. Les médicaments comme les inhibiteurs de l’enzyme de conversion de l’angiotensine (ECA), souvent utilisés pour réduire la charge de travail du cœur en cas d’insuffisance cardiaque, ne sont généralement pas utiles car ils réduisent trop la pression artérielle. Par conséquent, la quantité de sang qui circule dans le reste du corps est insuffisante. De même, la digoxine est généralement inutile et parfois nocive. Les agents bêta-bloquants sont mal tolérés par les personnes souffrant de cardiomyopathie restrictive et n’ont pas indiqué une capacité à améliorer la survie. Des anti-arythmiques peuvent être administrés pour éviter ou réduire les symptômes chez les personnes dont le rythme cardiaque est irrégulier.
Le trouble qui cause la cardiomyopathie restrictive peut parfois être traité afin d’éviter une aggravation de la lésion cardiaque ou même de l’inverser en partie. Par exemple, des saignées à intervalles réguliers réduisent la quantité de fer accumulée dans l’organisme en cas de surcharge en fer. Les personnes souffrant de sarcoïdose peuvent prendre des corticostéroïdes qui font disparaître le tissu granulomateux. Les corticostéroïdes peuvent aussi être utiles pour les troubles infiltrants éosinophiliques. Il n’existe toutefois aucun traitement pour de nombreux cas de cardiomyopathie restrictive.