Paris, France — Après des résultats positifs chez des patients atteints de sténose aortique ne pouvant être traités ni par chirurgie, ni par remplacement de la valve aortique par voie percutanée (TAVI), le traitement non invasif de la sténose cardiaque par ultrasons Valvosoft® va être évalué dans une nouvelle étude clinique, en vue d’obtenir le marquage CE, a annoncé la start-up française Cardiawave, fabricant du dispositif.
L’essai, qui a été autorisé par l’Agence nationale de sécurité du médicament et des produits de santé (ANSM), prévoit d’inclure une soixantaine de patients atteints de sténose aortique en France, en Allemagne et aux Pays-Bas. Ils seront suivis pendant un an pour vérifier l’absence d’effets indésirables graves et évaluer l’efficacité de cette approche innovante.
« Valvosoft® représente une solution thérapeutique non-invasive nouvelle, complémentaire au TAVI, qui permettra non seulement de traiter les patients les plus fragiles pour lesquels un TAVI n’est pas envisageable, mais également à plus long terme d’ouvrir la voie à de nouveaux patients atteints de rétrécissement aortique calcifié », précise la Pre Hélène Eltchaninoff (CHU de Rouen), coordonnatrice de l’étude au niveau national.
Ralentir l’évolution de la sténose
« Le traitement par ultrasons ne va pas remplacer le TAVI. L’idée est aussi de traiter les débuts de sténose aortique pour ralentir l’évolution vers un stade sévère, prendre en charge des patients plus jeunes et reculer ainsi au maximum l’intervention chirurgicale ou la pose d’un TAVI », a indiqué, auprès de Medscape édition française le Pr Emmanuel Messas (Hôpital Européen Georges-Pompidou, AP-HP, Paris), cardiologue et cofondateur de Cardiawave.
Cette nouvelle approche non invasive consiste à appliquer, de manière très précise et focalisée, un faisceau d’ultrasons au niveau des calcifications de la valve responsables de la sténose. « En baissant localement la pression hydrostatique, les ultrasons induisent la formation de bulles par le phénomène de cavitation, ce qui diminue la rigidité des tissus calcifiés et améliore l’ouverture de la valve », explique le cardiologue.
Développé par la start-up Cardiawave à partir des travaux du Laboratoire physique pour la médecine de Paris et de l’Institut Langevin (ESPCI/CNRS), l’appareil est placé au dessus du thorax du patient par un bras articulé. Le faisceau d’ultrasons, guidé par échographie, est appliqué et focalisé sur la valve par sessions de dix minutes pour un traitement de près d’une heure en ambulatoire.
Le phénomène de cavitation (formation et éclatement de bulles de vapeur sous l’effet d’une dépression) peut s’avérer douloureux, a précisé le Pr Messas. Le traitement se fait alors sous antalgique, voire sous sédation plus ou moins profonde, selon le profil des patients. Il faut également veiller au risque d’arythmie (extrasystole ou tachycardie ventriculaire) en cas de cavitation sur le myocarde.
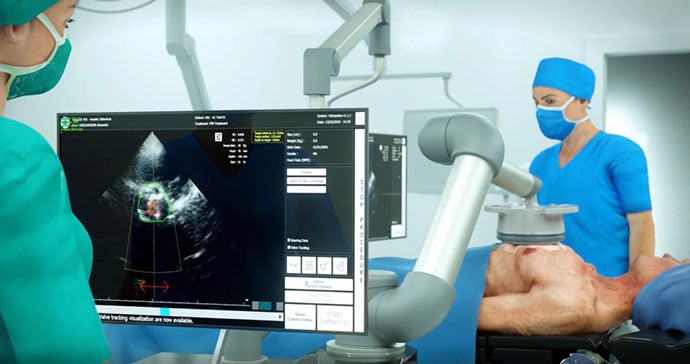
Absence de signes d’alerte
Après des essais sur l’animal, une première étude clinique a été menée sur 30 patients atteints de sténose aortique sévère pour évaluer la sécurité et la faisabilité du traitement. Ils étaient âgés en moyenne de 84 ans. « Chez ces patients, qui présentaient des valves très calcifiées et des comorbidités, la chirurgie et le TAVI étaient contre-indiqués ».
Les résultats préliminaires concernant dix patients ont été publiés. Un mois après la procédure, ils montrent une absence de mortalité, d’AVC et de tout autre événement indésirable grave lié au traitement. Une amélioration de l’état clinique a été observée chez six patients, avec une baisse du gradient de pression transaortique de 20 à 25%, tandis que la surface valvulaire a augmenté de 20%.
Une autre étude de faisabilité et de sécurité a été menée en parallèle au CHU de Belgrade en Serbie. L’évaluation, jusqu’à présent conduite chez 9 patients atteints de sténose aortique, a intégré un examen par IRM cérébral avant et après la procédure. « Aucune embolie cérébrale n’a été constatée après le traitement par ultrasons », indique le cardiologue.
L’ensemble des résultats de ces deux premiers essais cliniques vont être présentés ce mois-ci, lors du congrès de l’EuroPCR 2022, à Paris, a précisé le Pr Messas.
Une méthodologie à affiner
Après ces résultats positifs, l’ANSM a autorisé une nouvelle étude clinique visant à obtenir le marquage CE pour une version améliorée du dispositif, a expliqué le cardiologue. « Une deuxième version de la machine a été élaborée. Le nouveau modèle est plus performant et utilise l’échographie à biplan et non plus à monoplan pour localiser la valve », ce qui offre une meilleure précision.
L’inclusion des patients devrait débuter en mai. Il est prévu d’inclure des patients avec une sténose aortique moins sévère, notamment des patients plus jeunes, chez qui le TAVI n’est pas indiqué. Le protocole prévoit un suivi à un mois, à six mois et à 12 mois pour évaluer notamment l’impact du traitement sur la qualité de vie des patients.
Une fois le marquage CE obtenu, « les centres hospitaliers universitaires vont pouvoir s’approprier la machine et mener eux-mêmes leurs propres essais cliniques », dans l’objectif d’affiner les indications et la méthodologie de la thérapie. « A terme, on peut imaginer une approche plus automatisée, comme l’est la radiothérapie dans le traitement du cancer ».
Cardiawave prévoit également de mener des essais cliniques « d’ici la fin de l’année » aux Etats-Unis, dans des hopitaux de New-York.


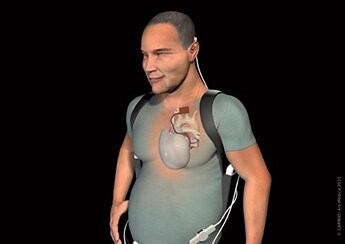 Le cœur artificiel total de CARMAT est composé d’une bioprothèse implantable et d’un système portable d’alimentation externe auquel elle est reliée en permanence. Il a été inventé par le Pr Alain Carpentier pour pallier le manque de greffons pour les personnes souffrant d’insuffisance cardiaque terminale irréversible. La première implantation chez l’homme a été réalisée en 2013 à l’hôpital européen Georges-Pompidou. En 2016, la startup française a cependant dû stopper son essai clinique après la mort d’un cinquième patient. Aussi, en 2018, la société a suspendu son essai en Europe en raison des risques de dysfonctionnement des prothèses. Après des modifications, le cœur artificiel a obtenu le « marquage CE » pour l’indication d’attente de transplantation, fin décembre 2020. En parallèle, l’ANSM a autorisé Carmat à reprendre les implantations en France en octobre dernier. Une nouvelle étude clinique chez 52 patients greffés a débuté.
Le cœur artificiel total de CARMAT est composé d’une bioprothèse implantable et d’un système portable d’alimentation externe auquel elle est reliée en permanence. Il a été inventé par le Pr Alain Carpentier pour pallier le manque de greffons pour les personnes souffrant d’insuffisance cardiaque terminale irréversible. La première implantation chez l’homme a été réalisée en 2013 à l’hôpital européen Georges-Pompidou. En 2016, la startup française a cependant dû stopper son essai clinique après la mort d’un cinquième patient. Aussi, en 2018, la société a suspendu son essai en Europe en raison des risques de dysfonctionnement des prothèses. Après des modifications, le cœur artificiel a obtenu le « marquage CE » pour l’indication d’attente de transplantation, fin décembre 2020. En parallèle, l’ANSM a autorisé Carmat à reprendre les implantations en France en octobre dernier. Une nouvelle étude clinique chez 52 patients greffés a débuté.